Tutorial 2: calculate the polarizability of CH4 molecule with Siesta
In this second tutorial, we will learn how to calculate the polarizability of a molecule using TDDFT as in tutorial 1. However in this case, we will use Siesta for the DFT calculations.
Requirements: have PyNAO, PySCF and Siesta installed
Run DFT with Siesta
We assume here that Siesta is properly installed and configured on your
system. We will assume also that siesta executable as been added to
your PATH
Method 1: run Siesta via the command line
The first way to run Siesta is to call the executable from the command line. For this, you should make sure to
to have a siesta
fdfinput file on the diskto have the necessary pseudopotential in the folder
First, we will define the siesta.fdf file (we already included the
pseudopotential files in the folder). In the siesta.fdf file
take note of the
SystemLabelvariable, you will need it for PyNAOthe option
COOP.Writemust be set toTruethe option
XML.Writemust be set toTrue
Let write the siesta.fdf file to disk
siestafdf = """SystemName siesta
SystemLabel siesta
COOP.Write True
DM.MixingWeight 0.01
DM.NumberPulay 4
DM.Tolerance 0.0001
MD.MaxForceTol 0.02 eV/Ang
MD.NumCGsteps 0
MaxSCFIterations 10000
PAO.BasisType split
SCFMustConverge False
WriteCoorXmol True
WriteDenchar True
XML.Write True
Spin non-polarized
XC.functional GGA
XC.authors PBE
MeshCutoff 3401.4232530459053 eV
PAO.EnergyShift 0.025 eV
NumberOfSpecies 2
NumberOfAtoms 5
%block ChemicalSpecieslabel
1 6 C
2 1 H
%endblock ChemicalSpecieslabel
%block PAO.BasisSizes
C DZP
H DZP
%endblock PAO.BasisSizes
AtomicCoordinatesFormat Ang
%block AtomicCoordinatesAndAtomicSpecies
0.000000000 0.000000000 0.000000000 1
0.629118000 0.629118000 0.629118000 2
-0.629118000 -0.629118000 0.629118000 2
0.629118000 -0.629118000 -0.629118000 2
-0.629118000 0.629118000 -0.629118000 2
%endblock AtomicCoordinatesAndAtomicSpecies
"""
with open("siesta.fdf", "w") as fl:
fl.write(siestafdf)
We can now launch DFT calculation with Siesta via the command line
!siesta <siesta.fdf > siesta.out
Method 2: run Siesta via ASE
The Atomic Simulation Environment (ASE) provides all an environment to run atomistic calculations. It is possible to run Siesta calculations via ASE, which is more friendly than running Siesta from the command line.
The advantage of using ASE is that you do not need to write the siesta
fdf input file by hand, and you don’t need to copy the
pseudopotentials. However, make sure that the ASE Siesta
calculator
is properly setup.
from ase.calculators.siesta import Siesta
from ase.units import eV, Ry
# First we need to build the molecule
# It can be done directly via ASE
from ase.build import molecule
atoms = molecule('CH4')
# Or you can load the molecule geometry from file
#import ase.io as io
#atoms = io.read(fname)
# set-up the Siesta parameters
siesta = Siesta(
mesh_cutoff=250*Ry,
basis_set='DZP',
pseudo_qualifier='gga',
xc="PBE",
energy_shift=(25 * 10**-3) * eV,
fdf_arguments={
'SCFMustConverge': False,
'COOP.Write': True,
'WriteDenchar': True,
'PAO.BasisType': 'split',
'DM.Tolerance': 1e-4,
'DM.MixingWeight': 0.01,
"MD.NumCGsteps": 0,
"MD.MaxForceTol": (0.02, "eV/Ang"),
'MaxSCFIterations': 10000,
'DM.NumberPulay': 4,
'XML.Write': True,
"WriteCoorXmol": True})
atoms.set_calculator(siesta)
# Run Siesta calculation
e = atoms.get_potential_energy()
print("DFT potential energy", e)
Run TDDFT with PyNAO
Now we can run TDDFT calculations similarly to what was done in tutorial 1 with PySCF.
The first step is to initialize the system and calculate the kernel by
calling tddft_iter.
For Siesta inputs, we need to call tddft_iter with the label
parameter. It must be the same label than the one indicated by
SystemLabel in the Siesta fdf file.
from pynao import tddft_iter
td = tddft_iter(label="siesta")
Once the kernel has been calculated, we can calculate the polarizability
of the molecule by calling the method comp_polariz_inter_Edir.
This method takes two main inputs:
freq: the frequencies at which to calculate the polarizability. It must be a complex number, the real part is the actual frequencies points, while the imaginary part is the actual broadening of the polarizability. In general it will betddft_iter.eps. The frequencies must be given in Hartree.Edir: the direction of the external electric field
import numpy as np
from ase.units import Ha
freq = np.arange(0.0, 25.0, 0.05)/Ha + 1j * td.eps
p_mat = td.comp_polariz_inter_Edir(freq, Eext=np.array([1.0, 1.0, 1.0]))
Total number of iterations: 10853
import matplotlib.pyplot as plt
h = 7
w = 4*h/3
ft = 20
fig = plt.figure(1, figsize=(w, h))
ax = fig.add_subplot(111)
ax.plot(freq.real*Ha, -p_mat[0,0, :].imag, linewidth=3)
ax.set_xlabel(r"Energy (eV)", fontsize=ft)
ax.set_ylabel(r"$P_{xx}$ (a.u.)", fontsize=ft)
fig.tight_layout()
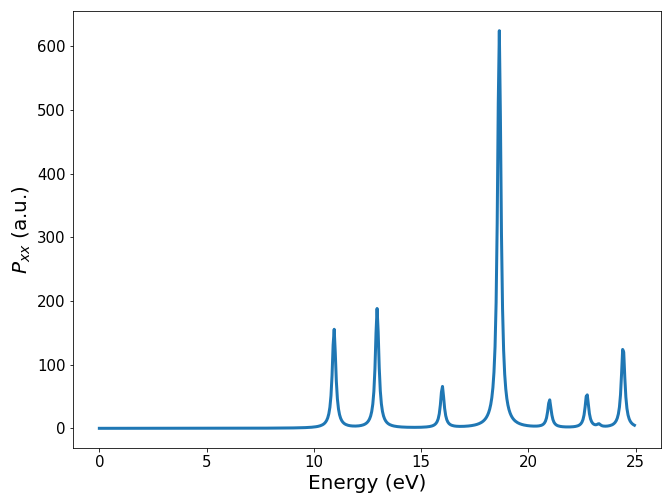
Remark
If you compare the spectra obtained in tutorial 1 and tutorial 2, you will see a quite different spectra, while the system is the same. Since the TDDFT part remained inchanged, it demonstrates how crucial is the quality of the DFT inputs in order to obtain reliable results.
The purpose of these tutorial is to demonstrate how to use PyNAO with PySCF and Siesta. It is the responsability of the user to ensure the quality of the calculations he is running.